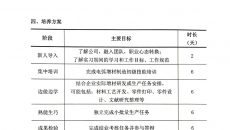
3D��ӡ�˹��wע�Ԍ���ָ��ԭ�t��������Ҋ�壩
һ��ǰ��
��ָ��ԭ�tּ�ڎ�����ָ����Ո�ˌ�3D��ӡ�˹��w�a(ch��n)Ʒע������Y���M�Мʂ䣬�ԝM�㼼�g���u�Ļ���Ҫ��ͬ�r�����ڌ��u�C����ԓa(ch��n)Ʒ�M�пƌWҎ(gu��)���Č��u����ߌ��u�������|����Ч�ʡ�
��ָ��ԭ�tϵ��3D��ӡ�˹��wע������Y�ϵ�һ��Ҫ����Ո��/���a(ch��n)��I(y��)������(j��)���w�a(ch��n)Ʒ�����Ԍ�ע������Y�ϵă�(n��i)���M�г䌍�ͼ�����������(j��)���w�a(ch��n)Ʒ�����Դ_�����еľ��w��(n��i)���Ƿ��m�ã������m�ã�����w�U�������ɼ������ĿƌW����(j��)��
��ָ��ԭ�t�nj���Ո��/���a(ch��n)��I(y��)�͌����ˆT��ָ�����ļ�����������ע�Ԍ������漰��������헣�����鷨Ҏ(gu��)���ƈ�(zh��)�У�������܉�M�����P��Ҏ(gu��)Ҫ�������������Ҳ���Բ��ã�������Ҫ�ṩԔ�����о��Y�Ϻ���C�Y�ϡ�������ѭ���P��Ҏ(gu��)�͘˜ʵ�ǰ����ʹ�ñ�ָ��ԭ�t��
��ָ��ԭ�t���ڬF(xi��n)�з�Ҏ(gu��)�͘˜��wϵ�Լ���ǰ�J֪ˮƽ���ƶ��ģ��S����Ҏ(gu��)�͘˜ʵIJ������ƣ��Լ��ƌW���g�IJ���l(f��)չ����ָ��ԭ�t���P��(n��i)��Ҳ���M���m�r���{(di��o)����
�����m�÷���
��ָ��ԭ�t�m�������w��׃���ߓp���M���w�г������c�����·������w���ںϹ̶���Ŀ�ĵ�3D��ӡ⁺Ͻ��˹��w�a(ch��n)Ʒ��
��ָ��ԭ�t�����Įa(ch��n)Ʒ����ü��������������ڵ�3D��ӡ���������ֶ����a(ch��n)�ģ���ϼ����o����(n��i)�̶�ϵ�y(t��ng)ʹ�õģ���������ֲ�ǵĘ˜ʻ�Ҏ(gu��)���TC4��TC4 ELI⁺Ͻ��˹��w�a(ch��n)Ʒ��
��ָ��ԭ�t���m���ڶ���ʽ��ȫ���p������Įa(ch��n)Ʒ�����m���ڿɓ��_�͡��Է�(w��n)�͡��ӑB(t��i)���߷��ںϵ��˹��w�a(ch��n)Ʒ��
���ڱ�ָ��ԭ�t��������3D��ӡ�˹��w���ɸ���(j��)�a(ch��n)Ʒ�ľ��w�OӋԭ�����Y���������������W���Լ��R��ʹ��Ҫ������ָ��ԭ�t�е����P��(n��i)�ݡ�
����ע������Y��Ҫ��
ע������Y�ϰ���ע������Y���ڰ��ա��P�ڹ����t(y��)����еע������Y��Ҫ��������C���ļ���ʽ�Ĺ��桷������ʳƷˎƷ�O(ji��n)���������ֹ���2014���43̖�����P�ڌ�ʩ�t(y��)����еע��������Ĺ��桷������ˎƷ�O(ji��n)�������ֹ���2019���46̖���M���ṩ�����������������������(n��i)�ݣ�
��һ���C���Y��
1.ע�Ԇ�Ԫ
ԓa(ch��n)Ʒһ���ɼ��������������ڵȼ��g�M���Ƃ䣬��ͬ���������췽ʽ����ͬ���ܵ�ԭ���Ϸ�ĩ�����֞鲻ͬ��ע�Ԇ�Ԫ��
�i���������a(ch��n)Ʒ�����鲻ͬע�Ԇ�Ԫ��
2.�a(ch��n)Ʒ����
�a(ch��n)Ʒ�������������a(ch��n)Ʒ���Q������e������a���Ƃ乤ˇ���a(ch��n)Ʒԭ���ϡ��A����;�����g����ָ�˼����ƶ�����(j��)�����ṩԔ���Įa(ch��n)Ʒ�Y���D���P�I�γߴ煢��(sh��)������K����|��Ļ��ȡ����w��λ���L���߶ȡ�ֲ�Ǵ��ߴ�ȣ����ṩ���OӋ����(j��)��
3.Ҏ(gu��)����̖
���ڴ��ڶ�N��̖Ҏ(gu��)��Įa(ch��n)Ʒ���ṩ�a(ch��n)Ʒ��Ҏ(gu��)����̖�Ą�������(j��)�����h����(j��)���w������(ji��)�ε��������ʽY�����R�����H��������g��·��ʽ�M�пƌW�Ěw���ͷ֙n���w�F(xi��n)����ͬ�΅���(sh��)�c������(ji��)�ε�ƥ���ԡ�
4.�a(ch��n)Ʒ���b
���_�a(ch��n)Ʒ�İ��bҎ(gu��)���b���ϡ������ʽ�͡���Ч���ޡ�
5.������ͬa(ch��n)Ʒ��ǰ���a(ch��n)Ʒ
��Ո�ˑ��C��ԓa(ch��n)Ʒ����(n��i)���о����R��ʹ�ìF(xi��n)��l(f��)չڅ�ݡ����ṩ����(n��i)��������ͬa(ch��n)Ʒ��ǰ���a(ch��n)Ʒ����Ϣ���U����Ոע�Ԯa(ch��n)Ʒ���аl(f��)������Ŀ�ġ����c���a(ch��n)Ʒ����ԭ�����Y���M�ɡ����칤ˇ��ԭ���ϡ�����ָ�ˡ��m�÷�������r�Č��ȡ�
6.�a(ch��n)Ʒ�m�÷����ͽ��ɰY
�ṩ�a(ch��n)Ʒ�m�÷�����������(ji��)�Σ����A��ʹ�íh(hu��n)�����A���c�����ʹ�õ���е��ʹ�÷��������g��ʽ���m����Ⱥ�����ɰY��Ϣ��
��������Y��
�a(ch��n)Ʒ���о��Y�ϑ����ļ��g����Փ�������a(ch��n)Ʒ���OӋ����(j��)�����g������ԭ�����x���ơ����a(ch��n)��ˇ���Ƽ���C���a(ch��n)Ʒ����ָ�˼��ƶ�����(j��)��������������C���a(ch��n)Ʒ���b��C���a(ch��n)Ʒ�����C���a(ch��n)Ʒ��Ч����C�ȣ����������������ڮa(ch��n)Ʒ���gҪ���е����P����ָ�ˣ������w��Ч�ԡ���ȫ��ָ���Լ��c�|���������Pָ�˵Ĵ_������(j��)�������õĘ˜�/�����Լ����õ����ɵȡ����ّ�������������������(n��i)�ݣ�
1.ԭ���Ͽ���
����⁺Ͻ��ĩ���ϑ�ԓ�ṩԔ���IJ��|�Σ�������ĩ�ɷ֡����ȡ������ֲ������ζȡ����b�ܶȡ����ܶȡ������Եȣ��������_�������ϵĘ˜ʡ���ԭ������ُ�������_ԭ���Ϲ����̲������Y�|�C���ļ������N�Pϵ�C���ļ������N�f(xi��)�h�����|���˜ʼ���C��档��Ո�ˑ�����ĩ�ɻ��մΔ�(sh��)�����f��ĩ��ϱ��������m�ϣ����M��Ҏ(gu��)�������ṩ�䌦��ӡ�^�̺ͮa(ch��n)Ʒ����Ӱ푵���C�Y�ϡ�
2.�a(ch��n)Ʒ�ɷֺ��@�M��Ҫ��
���_3D��ӡ�ĽK�a(ch��n)Ʒ���W�ɷ��Լ������ϵ����P�˜ʡ����_�@�M���c��ӡ����ӡλ�á����f��ĩ������֮�g���Pϵ��
3.�a(ch��n)Ʒ�^�Y����ȱ��
���ṩ�a(ch��n)Ʒ��ӡ��С��Ԫ����x���O������(j��)����϶�ʡ������z������(n��i)���Bͨ�ԡ���Y���ĺ�ȡ���϶�ݶȵ��x��_������(j��)�����������W���ܵ�Ӱ푡��������ɵ����á�����(n��i)���Y����ȱ����z�����ѡ��]�ȑ����ú��m���ֶ��M�Йz�y�����ƶ��ɽ��ܵ�ָ�˺��ṩ���P����(j��)����
4.�a(ch��n)Ʒ���W�����о�
��1���˜�ԇ�Ʌ����˜�YY/T 0959��YY/T0960�����P�˜��M���˹��w���o�B(t��i)���sԇ���s���С�Ť�Dԇ����ԇ����֮��߀��ԓ���]�˹��wÓ�������_���Ե�ԇԔ����������ָ�˼��z���Ĵ_������(j��)���ṩ���õ�ԭ����Փ���A���ṩ�漰�����о����Y�ϡ��īI�Y�Ϻ�/��˜��ı������ṩ�Y�����R���ɽ�������(j��)��
��2�������_��ԇ�ɿ��]�������Բ�����ʬ�w�����M���w���������Wԇ����C���a(ch��n)Ʒ���A�����ܡ��x�����ʬ�w���ܴ���ԓ�a(ch��n)Ʒ���m�÷���/�m���C���R��ʹ�����A�ڵĽ��ʲ�λ������W���������W���w��(n��i)�d�ɡ��c��(n��i)�̶���е���ʹ�á����g��·�ȷ��棬���ṩ�x�������(j��)��
��3���������Wģ�͜yԇ���ɿ��]����ע���˿��Բ��ü����������Wģ�͌��a(ch��n)Ʒ�M�����W������C�����ṩģ�ͅ���(sh��)�x�������(j��)��ԓ�������W�yԇĿ����Ҫ�����_ģ�M���w�����\�ӵ�Ҏ(gu��)�����ܼ��������W���|��׃�����d�ɼ����W���ĵĴ_����
��4�������r�x������(j��)�a(ch��n)Ʒ�m�ò�λ�x��ͬԇ���������������ʧЧ����̖Ҏ(gu��)���M�С����Բ�������Ԫģ�M�ȷ����M���x�����]�����H�R��ʹ���Ѓ�(n��i)�̶���е�������w�����������w�Ĺ������^�̌�����ģ���������ֲ�������Ԫ����ģ�ͅ���(sh��)��Ӱ푣����ṩ����Ԫģ�͜ʴ_�Ե���C�Y�ϡ���3D��ӡ�Įa(ch��n)Ʒ��ԓ���_�a(ch��n)Ʒ��ӡ��������(j��)������Ҏ(gu��)�����ϵ����W����(sh��)���ɲ���ͬ��ˇ�Ƃ�Ę�Ʒ�K�M�в�ͬ��������W�����о���
���W�yԇ����Б������c������ͬƷ�N�a(ch��n)Ʒ��(sh��)��(j��)��Ԕ������Փ�C�����Ȝyԇ���c���Ќ�(sh��)��(j��)���ȣ����Y����ֲ�빝(ji��)�ε����W���c���܇����o�����o��ʩ�������_�yԇ�Y���ɽ����Ե��ж�����(j��)��������ᘌ��Եć���(n��i)���ИI(y��)�˜ʻ�W�g�F�w�ٷ����R�е�ָ��Ҳ���Խ��ܡ�
5.���g�����о�
���������^���У����w��(j��ng)���ӶѯB���������ӟᡢ�������̵��^�̣��繤ˇ����(sh��)����̎���������a(ch��n)Ʒ�^���μӹ��Ĵ��ڽM���������Լ����������Ȳ������أ�ͬ�r��Y�����®a(ch��n)Ʒ�ȱ���e����������a(ch��n)Ʒ�����g���½������������ĺϽ�Ԫ���x�ӝ�����ߡ����hᘌ����g���ܰ����x�������M���о���ԇ��摪����Ԕ����ԇ����ԇ���|���ضȵȣ�������ɽ������M��Փ�C����ע���x�������r����ȱ���e���϶����ߡ���ӡ����λ�á����f��ĩ��ό��µĸ��g��Եȡ�
6.�����������u�r
�����¹�ˇ�Ͷ���µ��^��ıȱ���e���Ҵ�ӡ�^���з�ĩ�ɷֵĸ�׃�����������µ�����W�L�U����Ո����Ҫ��3D��ӡ�˹��w�a(ch��n)Ʒ�������������M���u�r�����h����(j��)GB/T16886ϵ�И˜ʽY�Ϯa(ch��n)Ʒ�����g�ԡ��x���������a(ch��n)Ʒ�������������M���u�r����ȱ�����P��(sh��)��(j��)�r�����M�б�Ҫ������������ԇ
7.��ԭ�ͼ�����(n��i)����
���]3D��ӡ��ˇ�ж�Y�������漰��ĩ��Ó���Լ�����ʬ�w�Ě�������Ҫᘌ���ԭ�ͼ�����(n��i)�����M����C��
8��MRI�����Ԝyԇ
����Ո�ˌ����a(ch��n)Ʒ�M����MRI�����Ե����P��C��������(j��)�о���棬�г�MRIԇ��O�䡢�ň����ȡ��������ʣ�SAR����ԇ��(sh��)��������λ��������Ӱ�u���Y��������Ո��δ�����a(ch��n)Ʒ�M��MRI�����Ե����P��C�������c���_ԓ�a(ch��n)Ʒ��δ�ڴŹ���(MRI)�h(hu��n)����ԓ�a(ch��n)Ʒ�Ĝ�������λ��r����Ӱ�M�Мyԇ�u���������f�����ľ�ʾ��ע�����P��(n��i)�ݣ���ʾ����ڵ��L�U��
9.�����ˇ�о�
�a(ch��n)Ʒ�轛(j��ng)��K��������_�����ˇ�������ͅ���(sh��)���͟o�����Cˮƽ��SAL����SAL���_��10-6���ṩ����_�J��档�����]�a(ch��n)Ʒ�ĸ߿�϶�ʺͱȱ���e������ؓ�d��Ӱ푡�����ʹ�õķ������׳��F(xi��n)����,�����_��������Ϣ����ȡ��̎�����������ṩ�о��Y�ϡ�
10.�a(ch��n)Ʒ��Ч�ںͰ��b�о�
���ա��oԴֲ�����t(y��)����е؛�܉�������Y��ָ��ԭ�t���ṩ�a(ch��n)Ʒ��Ч�ڵ���C��棬��ͬ���b�Įa(ch��n)Ʒ��քe�ṩ��C�Y�ϡ�����Ո���ṩ�����t(y��)����е�a(ch��n)Ʒ��؛����Ч����C�Y�ϣ��t���ṩ���c�������a(ch��n)Ʒ��ԭ���ϡ����������������������b���ϡ����b��ˇ�����b��ʽ������Ӱ�������ܵ����ط�����е�ͬ�Ե��C���Y�ϡ�
11.���
���ա��Q���Ƿ��_չ�t(y��)����е�R��ǰ�����о��ļ��g����ָ��ԭ�t������o��ͨ�^�c�����Юa(ch��n)Ʒ�Ķ�Y�������M�е�ͬ��Փ�C����ᘌ�3D��ӡ�˹��w�a(ch��n)Ʒ���ض�Y���M�Є�������c�M���a(ch��n)Ʒ�A����;�������Ľ��ʲ�λ�Ą���ģ���M����C���Է������a(ch��n)Ʒ���¹��γɡ����L����ȡ��Լ��������������W����ָ�ˣ��������Y����ȡ���ںϲ�λ�M���w���������Wԇ��u�r��Y�Ϗ��ȡ���Ӷ��Լ����s���ȵȣ��ȵ�Ӱ푣�ͬ�r�u�r�x���������܇��M����Ӱ푡���ͨ�^�����L��ĕr�g�ͽY�����M�к������_���R��ԇ����A�ڹ��L��ĕr�g�ͽY�������Ը��õĴ_���R��ԇ��^�y�r�g������ګ@�Ì�����ʹ���S���C�ęC���M�У����@�������������팏��ͱO(ji��n)�ܡ�
���������a(ch��n)������Ϣ
Ԕ���a(ch��n)Ʒ�����a(ch��n)�^�̣��ṩ���a(ch��n)��ˇ���̈D�����_�����^�̺��P�I��ˇ�����U�����^�̿����c�����ƅ���(sh��)�������a(ch��n)��ˇ�Ŀɿ��ԡ���(w��n)���ԑ��M�д_�J��
1��ӡ��ˇ��C
���_3D��ӡœ�ҭh(hu��n)���Լ����ϳ����P�I����(sh��)���������x�Ĺ�ˇ����(sh��)���a(ch��n)Ʒ��ӡ����λ�á�֧�νY�����M����C�����C�a(ch��n)Ʒ���ܵ�һ���ԡ�
2��̎��������C
���_�a(ch��n)Ʒ3D��ӡ�ĺ�̎����ʽ�������o����ȥ֧�Ρ������ĩ��ϴ�ȣ������u����̎����ˇ�����ϺͽK�a(ch��n)Ʒ�İ�ȫ����Ч�Ե�Ӱ푡��ɲ�����ͬ��ˇ����(sh��)�Ƃ�ĘӉK�M����C��
3��ϴ��ˇ��C
���_ԭ���ϼ����a(ch��n)��ˇ���漰�ĸ��N�ӹ��������|�����Ƙ˜ʡ����_�a(ch��n)Ʒ����ϴ�^�̣��ṩ��(j��ng)��ϴ�^�̺�ӹ������������Ƶ���C�Y�ϡ�
4���ơ����a(ch��n)����
��ԓ�����ơ����a(ch��n)���صČ��H��r�����ж������ơ����a(ch��n)���أ�����ÿ�����ơ����a(ch��n)���صČ��H��r�M�и�����
���ģ��a(ch��n)Ʒ���L�U�����Y��
����(j��)YY/T 0316���t(y��)����е�L�U���팦�t(y��)����е�đ��á�������R�e3D��ӡ�˹��w���OӋ��ԭ���ϡ����a(ch��n)�ӹ������b��������\ݔ���A�桢ʹ�õ��������ڃ�(n��i)�����h(hu��n)��(ji��)�İ�ȫ����,������WΣ�����h(hu��n)��Σ�������Pʹ�õ�Σ��������ʧЧ���ϻ����惦���������Σ���ȷ��棬���a(ch��n)Ʒ�M��ȫ����L�U��������Ԕ������ȡ���L�U���ƴ�ʩ��
�ṩ�a(ch��n)Ʒ����ǰ�����L�U��������M��ȫ���u�����γɵ��L�U�����棬�ˈ��ּ���f�������Z�L�U����Ӌ���ѱ��m��?sh��)،�ʩ���C��ʣ���L�U�ǿɽ��ܵģ�����ǡ��?sh��)ķ����@�îa(ch��n)Ʒ���P�����S����ͨ���R�����õ���Ϣ��
�L�U�����摪�����L�U�������L�U�u�r���L�U���ƵȮa(ch��n)Ʒ�L�U���������P�Y�ϣ����ّ������a(ch��n)Ʒ��ȫ������Ρ��a(ch��n)Ʒ���AҊ��Σ����Σ��������Σ��f��Σ�������AҊ�¼����У���Σ�������������Σ��̎���Ϳ��ܰl(f��)���ēp��֮�g���Pϵ���L�U�u�r���L�U���ƴ�ʩ�Լ�ʣ���L�U�u�r�R������
���壩�a(ch��n)Ʒ�ļ��gҪ��
�a(ch��n)Ʒ���gҪ���ա��t(y��)����е�a(ch��n)Ʒ���gҪ��ָ��ԭ�t���M�о�������Ո�ˑ��Y�Ϯa(ch��n)Ʒ�ļ��g�������R��ʹ����r���_���a(ch��n)Ʒ��ȫ��Ч���|���ɿصļ��gҪ���c�z�����a(ch��n)Ʒ���gҪ���Б����_Ҏ(gu��)����̖���䄝�ֵ��f�����a(ch��n)Ʒ����ָ�˼�ԇ�����a(ch��n)Ʒ����һ����Ϣ��ԭ���ϡ��M�ɳɷ֡��Y���ȣ����a(ch��n)Ʒ�����ʽ��؛����Ч�ڡ��a(ch��n)Ʒ���gҪ���еă�(n��i)�����Ç��Ҙ˜ʡ��ИI(y��)�˜ʻ��Ї�ˎ��ģ������C����Ч�ԣ���ע�������˜ʵľ�̖����̖���Ї�ˎ��İ汾̖��
���wָ�˰���������������(n��i)�ݣ�
1. �a(ch��n)Ʒ��̖/Ҏ(gu��)�����f����
2.���W�ɷֺ��@�M�������_��ĩ���ϡ��˹��w�a(ch��n)Ʒ�Ļ��W�ɷ֡����_3D��ӡ�˹��w���@�M�������������_��ӡ����͟�̎���c�@�M�����Pϵ��
2.�^�Y�������_3D��ӡ�Ŀ����z������϶�ʡ�ͨ���ʡ���Ӻ�ȵȡ�
3.�����|������ײ��ֱ��摪�o����Ƥ��Ҳ���o�Ƕ��K�ӹ����e���������Ⱦ���Ӳ����Д�z�F(xi��n)������⡣
4.��(n��i)��ȱ�ݡ�������(n��i)���Y����ȱ����z�����ѡ��]���M�Йz�y�����ƶ��ɽ�������(j��)��
5.���W���ܡ���Ҏ(gu��)���a(ch��n)Ʒ��Ӳ�ȡ����ȡ��o�B(t��i)Ť�D�����s�����С����ݡ�Ó�������W����ָ�ˡ�
6.���o����
7.������(n��i)����
�����Q�������������g����(sh��)���ܣ������ڮa(ch��n)Ʒ���gҪ��������Ҏ(gu��)����
�������a(ch��n)Ʒ��ע�ԙz���
��Ո�ˑ��ṩ�����t(y��)����е�z��Y�|���t(y��)����е�z�C�����ߵęz�����A�u�r��Ҋ�����⣬߀���ṩ�z��ƷҎ(gu��)����̖���x������(j��)��
���z���̖�a(ch��n)Ʒ�����DZ�ע�Ԇ�Ԫ��(n��i)�܉��������������̖�a(ch��n)Ʒ��ȫ�Ժ���Ч�Եĵ��ͮa(ch��n)Ʒ��
���ߣ��R���u�r
���ա��t(y��)����е�R���u�r���gָ��ԭ�t�����������t(y��)����е�����R��ԇ(sh��)��(j��)���gָ��ԭ�t���Լ�������ֲ�����R���u�r�|������ע�Լ��g����ָ��ԭ�t��������Ҋ�壩���ύ�R���u�r�Y�ϡ��漰���R���u�r���{������Ո�m�÷����ĸ���λ�����i���������ȡ����c���R��ԇ��Լ��cͬƷ�N�����д����Ɇ��ģ���Ҫ�M���R��ԇ��M����C���R���^��r�g������6���£������ٰ�������(n��i)�ݣ����˹��wֲ����ߵ���ʹ�����ܵ��u�������w�ں��ʡ����w���ȡ��ں϶����ȡ��w�߶ȵ�Ӱ��W�Ĝy�u���Լ��a(ch��n)ƷÓ�������ݡ����ѵȲ����¼���Ԕ������ӛ䛡�
���ˣ��a(ch��n)Ʒ�f�����͘˺�
�a(ch��n)Ʒ�f�����͘˺������ϡ��t(y��)����е�f�����͘˺�����Ҏ(gu��)����������ʳƷˎƷ�O(ji��n)�������������6̖����Ҫ��
�a(ch��n)Ʒ�R���m�÷���/�m���C�����ɰY��ע����헑��c�R��ԇ���R���u�r����C�ķ���һ�¡�
�a(ch��n)Ʒ��Ч�ڡ����õĜ�����������]���õĜ����������Ϣ���c�a(ch��n)Ʒ���g�������һ�¡�
�ġ������īI
1�����t(y��)����е�O(ji��n)������l���������A���͇�����Ժ���680̖��
2�����t(y��)����еע�Թ����k����������ʳƷˎƷ�O(ji��n)�������������4̖��
3�����t(y��)����е�f�����͘˺�����Ҏ(gu��)����������ʳƷˎƷ�O(ji��n)�������������6̖��
4�����t(y��)����е�R��ԇ��|������Ҏ(gu��)����������ʳƷˎƷ�O(ji��n)�������������25̖��
5.���t(y��)����е�R���u�r���gָ��ԭ�t��������ʳƷˎƷ�O(ji��n)����������ͨ��2015���14̖��
6.���P�ڹ����t(y��)����еע������Y��Ҫ��������C���ļ���ʽ�Ĺ��桷������ʳƷˎƷ�O(ji��n)���������ֹ���2014���43̖��
7. GB/T 16886���t(y��)����е����W�u�r��ϵ�И˜�
8. YY/T 0959������ֲ���� �g�ں������W����ԇ����
9. YY/T 0960������ֲ���� �g�ں����o�B(t��i)�S�s����ԇ����
10. ASTM WK60265 New Guide for Assessing the Removal of Additive Manufacturing Residues in Medical Devices Fabricated by Powder Bed Fusion [2019-08-07].
�塢��݆�λ
����ˎƷ�O(ji��n)���������t(y��)����е���g���u����











 �ղ�
�ղ� �D��
�D�� ֧��
֧�� ����
����
 �����W(w��ng)����11010802043351
�����W(w��ng)����11010802043351